Publish: 2024.01.02. / Nature methods
AlphaFold나 RosseTTAFold는 "genetic database 기반 MSA 데이터" 와 "structure database 기반 pair representation" 간의 정보를 교환하고, iterative하게 받아올 수 있도록 model architecture를 구성하여 좋은 성능을 보였다. 본 논문의 저자들 또한 DeepMSA2라는 모델에서 비슷한 concept을 가지고, genomic and metagenome sequence database를 기반하여 모델을 만들었으며 CASP15 데이터로 비교해본 결과, AlphaFold2-Multimer server (v.2.2.0) 보다 더 좋은 성능을 보였다고 한다.
DeepMSA2는 크게 2가지 pipeline으로 구성되어 있다.
1. Monomer MSA construction

1) Iterative Multiple sequence alignments(MSA) generation step
Three parallel blocks
(1) dMSA
- Modified from MSA generation tool, DeepMSA
- Generates up to 3 MSAs by a three-stage procedure (HHblits / Jackmmer / HMMsearch)
- Iteratively search the genomic and metagenomics sequence databases
- Neff value >128 이면 stop (most three MSAs will be generated)
- HHblits : Search Uniclust30 (parameters: -diff inf -id 99 -cov 50 -n 3)
- Jackmmer : Search Uniref90 (parameters: -N 3 -E 10 --incE 1e-3) -> Potentially homologous seqences 선정 위함
- HMMsearch : Search HHM against Metaclust metagenome DB (parmeters: -E 10 --incE 1e-3)
- BLAST filter (PSIBLAST): Speed up thd custom DB and filter out noisy raw sequences (rank the homolgous relation up to 30,000) -> filter가 없던 AlphaFold2 보다 성능이 좋았던 이유로 생각됨
- By kClust, Jachmmer로부터 얻어진 full-length sequences clustering into sequence clusters (30 % seqence identity cutoff)
- By Clustal Omega, realign sequences within each cluster into aligned sequence profiles
- Aligned sequence profiles: By using the 'hhblitdb.pl' script from the HH-suite package, HHblits-style DB로 convert -> 다시 stage1의 HHblits에 들어감 (그래서 iterative)
- MSA: By HHMbuild from the HMMER package, HMM(hidden Markov model)으로 convert
(2) qMSA
- HHblits (v.2)
- Jackhmmer
- HHblits (v.3)
- HMMsearch : Search aganist [Uniref30, Uniref90, BFD, Mgnify DB]
(3) mMSA
- HMMsearch (parameters: -E10 --incE 1e-3)
- Search through a metagenomics DB combining JGIclust (Joint Genome Institute), TaraDB, MetaSourceDB
- Used as the target DB -> serached by HHblits (v.2) with three seed MSAs (MSAs from dMSA stage2 / qMSA stage 2 / 3)
2) Deep learning-based ranking step
- Confidence scores of predicted structure models
Final MSA selection
- MSAs generation step에서 MSAs 10개 선정
- 각 MSAs는 modified AlphaFold2를 거쳐 5개의 구조 생성
- 생성된 5개 구조 중, 가장 높은 pLDDT score를 가진 것이, 해당 MSA의 rank score가 됨
- 10개의 MSA 중 가장 높은 rank score을 가진 것이, final selected MSA
2. Multimer MSA construction

1) Monomeric MSA generation
- DeepMSA2-Monmer pipeline과 동일
- 하지만 top-ranking 1개만 사용한 monomer와는 달리, 각 chain에 대해 10개의 MSAs 사용됨
- 다른 component chains들 간의 quaternary orientations modeling에 사용
2) MSA pairing
Two types of complexes
(1) Homomeric complexes
- all of the monomer MSAs
- top M MSAs are selected for each monomer chain so that M^N(complex내 distinct chains 수) distinct paired MSAs can be created for the complex (maximal M^N <= 100)

(2) Multimeric MSAs
- Created by concatenating each of the monomer MSAs n(monomer chains 수) times side-by-side
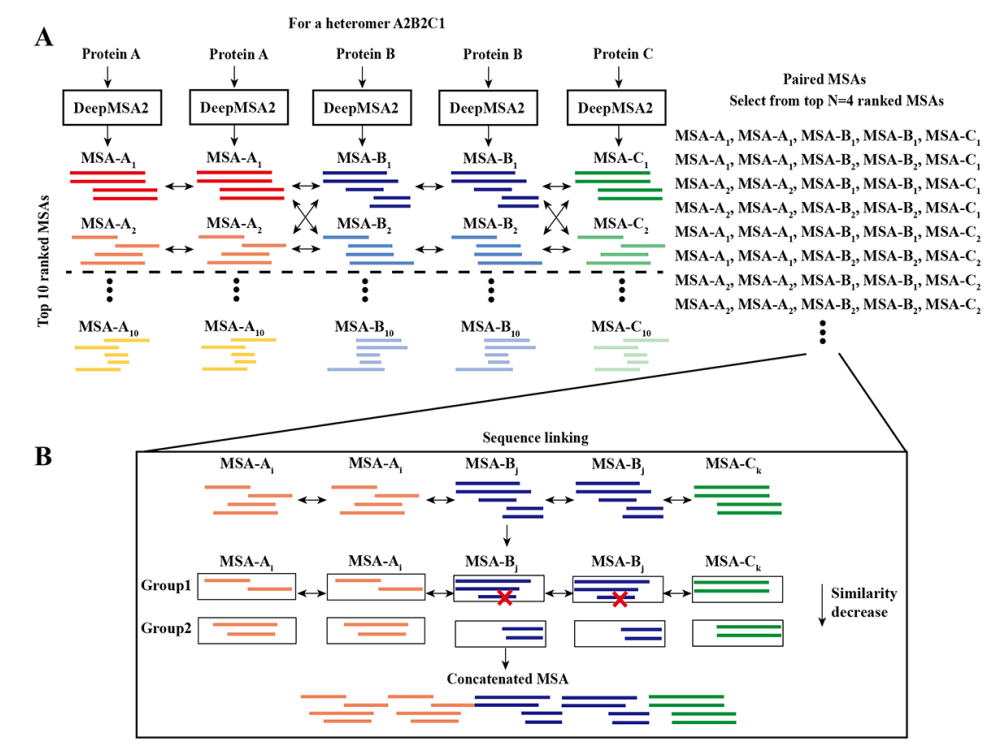
3) Sequence linking
M^N개의 monomeric MSAs
- monomeric MSAs: concatenated into a multimeric MSA
1. Based on the UniProt annotated species, the sequences in each monomeric MSA are grouped
2. Based on the sequence identity to the query sequence, the sequences in earch group are orderd
4) Concatenated MSA selection
- M-score를 기준으로 M^N concatenated MSAs 중 top 25개 선정
- Neff (the number of effective sequences)


- Neff: the depth of the concatenated MSA calculated
- L: the length of the query sequence
- N: the number of sequences contained in the MSA
- Sm,n: the sequence identity between the mth and nth sequences